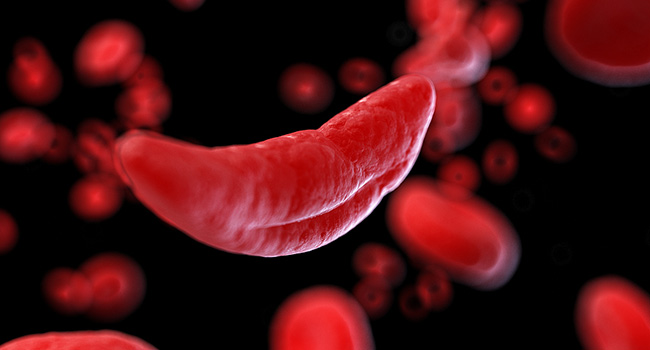
Sickle cell disease (SCD) is a group of hereditary blood disorders of which the most common type is sickle cell anemia. SCD affects the hemoglobin in red blood cells, the component of red blood cells that carry oxygen in the body. People with SCD have an abnormality in their hemoglobin which causes the shape of the red blood cells to distort into a crescent or a sickle shape, affecting the red blood cells’ longevity and ability to transport oxygen around the body. It is most common amongst people whose ancestors came from Africa, and the most common inherited blood disorder in the United States.
Risk Factors for Sickle Cell Disease
Sickle cell disease is inherited. For a person to have sickle cell, they must inherit one copy of the sickle cell gene from each parent. The following factors increase the likelihood of SCD:- Parents’ genetics: If both parents have one copy each of the sickle cell gene, there is a 1 in 4 chance of the child being born with SCD. There is a 1 in 2 chance of the child being a carrier as well, and a 1 in 4 chance of the child not having SCD or being a carrier either. If both parents have SCD, the child will have SCD. If only one parent has SCD, the child will be a carrier.
- Ethnic background: Most people with SCD or with one copy of the sickle cell gene are of sub-Saharan African descent. Those people whose ancestors are from South America, Cuba, Central America, Saudi Arabia, India, Turkey, Greece, and Italy are also at risk of inheriting SCD.
Clinical Features of SCD
Symptoms of SCD generally begin in early childhood, at as young as 4-5 months old, although the severity of the symptoms can differ from person to person. Sickle cell disease can present with the following symptoms:- Anemia: Because sickle cells have a much shorter lifespan than normal red blood cells, patients with SCD can have a scarcity of red blood cells and develop anemia.
- Fatigue: The lack of red blood cells means less oxygen is carried throughout the body. This can lead to chronic fatigue.
- Episodes of pain, or pain crises: Sickle cells cannot pass through blood vessels as easily as red blood cells; this can cause blockage of blood flow. Pain can occur in chest, abdomen, joints, and even bones. The intensity and length of the pain can vary, ranging from a few hours to a few weeks. Some patients only experience a few pain crises a year, while it may be more frequent in others. A severe pain crisIs might result in the need for hospitalization.
- Swelling of hands and feet
- Delayed growth or puberty
- Frequent infections
- Vision problems
Diagnosing Sickle Cell Disease
A simple blood test can determine the presence of SCD by detecting the abnormal hemoglobin. In countries like the US and UK, newborns are routinely screened for sickle cell disease. Adults can also get this test if they are concerned about being carriers of the sickle cell gene. The screening test can only determine whether the sickle cell gene exists, and a positive test does not necessarily mean that the person has SCD. Further testing is then required to determine whether the person is a carrier or has SCD.A test for SCD can also be performed before birth using a small sample of amniotic fluid or the placenta.
Treatment and Management of Sickle Cell Disease
The only known cure for SCD is a stem cell or bone marrow transplant, but these are not done frequently because of the significant risk involved. Where possible, SCD is managed through dietary supplements such as folic acid, which stimulates red blood cell production. If sickle cell anemia is particularly persistent or severe, blood transfusions may be necessary. For other complications that arise due to SCD, various treatment options are available:- Hormonal medicine for delayed puberty
- Painkillers to manage pain crises
- Gallbladder removal surgery for gallstones
- Leg ulcers can be treated by cleaning and dressing the ulcers